Polycystická nemoc ledvin: Porovnání verzí



(přepsání článku) |
m (preklep) |
||
| Řádek 30: | Řádek 30: | ||
Diagnóza se určuje u pacientů, kteří zaznamenávají výskyt choroby v rodině, nachází se u nich mírná [[proteinurie]] nebo mikro-/makrohematurie. Diagnózu lze určit pomocí USG, [[vylučovací urografie]] nebo [[CT]]. Při nálezu defektního [[gen]]u je taktéž diagnostikována ADPKD. | Diagnóza se určuje u pacientů, kteří zaznamenávají výskyt choroby v rodině, nachází se u nich mírná [[proteinurie]] nebo mikro-/makrohematurie. Diagnózu lze určit pomocí USG, [[vylučovací urografie]] nebo [[CT]]. Při nálezu defektního [[gen]]u je taktéž diagnostikována ADPKD. | ||
Pro tuto chorobu neexistuje [[kauzální terapie]]. Je zde léčena chronická renální insuficience, | Pro tuto chorobu neexistuje [[kauzální terapie]]. Je zde léčena chronická renální insuficience, hypertenze a močové infekce. | ||
Verze z 9. 11. 2018, 15:33
Polycystická nemoc ledvin je onemocnění charakterizované výskytem cyst v ledvinné kůře a/nebo parenchymu. Přítomnost cyst způsobuje úbytek funkčního parenchymu ledviny a může rezultovat v renální insuficienci. Na základě dědičnosti můžeme chorobu rozdělit na autosomálně dominantní a autosomálně recesivní.
Autosomálně dominantní polycystická nemoc ledvin
| Polycystická choroba ledvin (AD) | |
 Průřez polycystickou ledvinou | |
| Klinický obraz | bolest v bedrech, hematurie, močové infekce |
|---|---|
| Příčina |
mutace genu PKD1 (AD) 16. chromozóm |
| Diagnostika | CT, USG |
| Incidence ve světě | 1/1 000 |
| Prognóza | renální selhání |
| Klasifikace a odkazy | |
| MKN-10 | Q60-Q64 |
| MeSH ID | D016891 |
| OMIM | 173900 |
| orphanet | ORPHA730 |
| MedlinePlus | 000502 |
| Medscape | 244907 |
Autosomálně dominantní polycystická choroba ledvin (ADPKD) je nejčastější vrozená renální choroba (incidence 1:1000). Nacházejí se zde četné korové a dřeňové cysty v obou ledvinách narůstají a vedou k redukci funkčního parenchymu.
Nejčastěji se jedná o mutaci genu PKD1 na 16. chromozómu (90 %).
Tato nemoc probíhá ve většině případů bezpříznakově, často se jedná o náhodný nález při USG. Příznaky se objevují v podobě bolesti beder nebo břicha, hematurie, infekce močových cest, nefrolitiázy, hypertenze, jaterních cyst, prolapsu mitrální chlopně, interkraniálního aneuryzmatu nebo postupného zhoršování renálních funkcí. Výše uvedené příznaky se dostavují až ve středním věku.
Diagnóza se určuje u pacientů, kteří zaznamenávají výskyt choroby v rodině, nachází se u nich mírná proteinurie nebo mikro-/makrohematurie. Diagnózu lze určit pomocí USG, vylučovací urografie nebo CT. Při nálezu defektního genu je taktéž diagnostikována ADPKD.
Pro tuto chorobu neexistuje kauzální terapie. Je zde léčena chronická renální insuficience, hypertenze a močové infekce.
Autosomálně recesivní polycystická nemoc ledvin
| Polycystická choroba ledvin (AR) | |
 Polycystické ledviny | |
| Klinický obraz | oligohydroamnion, rychlý rozvoj renální insuficience v poporodním období |
|---|---|
| Příčina |
mutace genu PKDH1 (AR) 6. chromozóm |
| Diagnostika | prenatální sonografie od 24. týdne těhotenství |
| Incidence ve světě | 1/20 000–40 000 |
| Prognóza | renální selhání |
| Klasifikace a odkazy | |
| MKN-10 | Q60-Q64 |
| MeSH ID | D017044 |
| OMIM | 263200 |
| orphanet | ORPHA731 |
| MedlinePlus | 000502 |
| Medscape | 983281 |
Autosomálně recesivní polycystická choroba ledvin (ARPKD) je podstatně vzácnější, než její autosomálně dominantní forma (incidence 1:20 000–40 000).
Je způsobena genovou mutací na 6. chromozómu.
Jedná se o rychlý rozvoj renální insuficience v poporodním období. U pacientů s touto chorobou se projevuje hypoplázie plic, oligohydramnion nebo fibróza jater s rozvojem portální hypertenze.
Nemoc je diagnostikována pomocí USG ledvin, prenatální stanovení dg. možné již od 24. týdne gravidity.
Při této diagnoze závisí na srávném zvádnutí období po porodu. Později nastává terapie CHSL, případně jaterního selhání.
Galerie
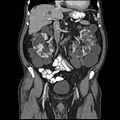
CT polycystické choroby ledvin

MRI obraz polycystické choroby ledvin
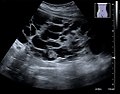
USG zobrazení polycystické ledviny



Použitá literatura
- DÍTĚ, P., et al. Vnitřní lékařství. 2. vydání. Praha : Galén, 2007. ISBN 978-80-7262-496-6.
