Polycystická nemoc ledvin
(přesměrováno z Polycystická choroba ledvin/autosomálně dominantní)


Polycystická nemoc ledvin je onemocnění charakterizované výskytem cyst primárně v ledvinné kůře. Na základě dědičnosti můžeme chorobu rozdělit na autosomálně dominantní a autosomálně recesivní.
Autosomálně dominantní polycystická nemoc ledvin (ADPKD)[upravit | editovat zdroj]
| Polycystická choroba ledvin (AD) | |
 Průřez polycystickou ledvinou | |
| Klinický obraz | bolest v bedrech, hematurie, močové infekce |
|---|---|
| Příčina |
mutace genu PKD1 (AD) 16. chromozóm |
| Diagnostika | CT, USG |
| Incidence ve světě | 1/1 000 |
| Prognóza | renální selhání |
| Klasifikace a odkazy | |
| MKN-10 | Q61.2 |
| MeSH ID | D016891 |
| OMIM | 173900 |
| orphanet | ORPHA730 |
| MedlinePlus | 000502 |
| Medscape | 244907 |
Je nejčastější vrozená renální choroba (s výskytem 1:1 000 až 1:400). Nacházejí se zde četné korové a dřeňové cysty, které rostou a vedou k redukci funkčního parenchymu.
Geneticky jde o heterogenní onemocnění, rozlišujeme gen PKD1 (se závažnějším průběhem) a PKD2 (s mírnějším průběhem). Genovým produktem je protein polycystin, který má souvislost s poruchou řasinek a proto většina polycystických chorob ledvin je dnes řazena mezi ciliopatie. Přesný mechanismus vzniku cyst však není znám.
Klinika: dominujícím klinickým projevem ADPKD je postižení ledvin. USG prokazuje mnohočetné oboustranné makrocysty prostupující renální parenchym již během dětství, vzácně už v kojeneckém věku. Ledviny jsou zvětšené.
Onemocnění běžně provází:
 Bolest břicha (u 50% dětí)
Bolest břicha (u 50% dětí)- Hypertenze, arteriální
- Proteinurie (mikroalbuminurie u 30-60%)
- Abdominální rezistence při somatickém vyšetření
- Snížení koncentrační schopnosti (u 50%)
Extrarenální příznaky (méně časté):
- Cysty v játrech, pankreatu, ovariích, slezině či mozku
- Chlopenní srdeční vady (nejčastěji prolaps mitrální chlopně)
- Aneurysmata intrakraniálních arterií (u všech dětí s pozitivní anamnézou aneurysmat nebo bolestí hlavy je doporučen screening aneurysmat MR angiografií)
Dalšími přidruženými nálezy mohou být hernie, bronchiektázie, aortální nebo koronární aneurysmata. V projevech onemocnění existuje velká variabilita, jak ve věku při manifestaci postižení, tak ve stupni postižení. Nelze tak předpovědět průběh nemoci u dítěte na základě průběhu u jeho rodiče či prarodiče.
Diagnostika: výskyt choroby v rodině, proteinurie nebo mikro-/makrohematurie.
Diagnózu lze určit pomocí USG, vylučovací urografie nebo CT a rodinné anamnézy. Při nálezu defektního genu je taktéž diagnostikována ADPKD.
Diagnostickým kritériem pro ADPKD u dětí je přítomnost alespoň 3 renálních cyst ve spojení s pozitivní RA polycystózou nebo přítomnost již 2 cyst při známé mutaci.
Terapie: léčba hypertenze (cílem je krevní tlak < 120-125/80 mmHg), redukce proteinurie. V rámci moderních postupů se v klinických studiích nadějně jeví užití antagonistů Rp pro ADH, tzv. vaptanů a analog somatostatinu. Protože nejsou pro léčbu ADPKD oficiálně uznány, je snaha o léčbu „vodou“, tj. navýšení příjmu tekutin tak, aby sekrece ADH byla co nejnižší (to představuje U-osmo < 300 mosmol/kg).
Komplikace: nejčastější „očekávanou“ komplikací je chronické renální selhání, nejčastěji ve 3.-4. deceniu, recidivující IMC, abrupce aneurysmat.
Prognóza: prognosticky je nejzávažnějším faktorem věk manifestace (onemocnění má nejtěžší průběh již během 1. roku života - „very early onset ADPKD“ (VEO-ADPKD).
Autosomálně recesivní polycystická nemoc ledvin[upravit | editovat zdroj]
| Polycystická choroba ledvin (AR) | |
 Polycystické ledviny | |
| Klinický obraz | oligohydroamnion, rychlý rozvoj renální insuficience v poporodním období |
|---|---|
| Příčina |
mutace genu PKDH1 (AR) 6. chromozóm |
| Diagnostika | prenatální sonografie od 24. týdne těhotenství |
| Incidence ve světě | 1/20 000–40 000 |
| Prognóza | renální selhání |
| Klasifikace a odkazy | |
| MKN-10 | Q61.1 |
| MeSH ID | D017044 |
| OMIM | 263200 |
| orphanet | ORPHA731 |
| MedlinePlus | 000502 |
| Medscape | 983281 |
Je podstatně vzácnější vrozená renální choroba (s výskytem 1:40 000 až 1:10 000) a je způsobena genovou mutací na 6. chromozomu (6p12.2).
Projevuje se rychlým rozvojem renální insuficience v poporodním období. U dětí s touto chorobou se v brzkém věku vyskytuje hypoplázie plic, oligohydramnion nebo fibróza jater s rozvojem portální hypertenze.
Nemoc je diagnostikována pomocí USG ledvin, prenatální stanovení diagnózy je možné již od 24. týdne gravidity.
Při této diagnoze závisí na správném zvládnutí období po porodu. Později nastává terapie CHSL, případně jaterního selhání.
Genetika: AR heredita, porucha genu polycystic kidney and hepatic disease 1 (PKHD1), který kóduje protein fibrocystin.
Patologie: onemocnění postihuje 2 orgány, ledviny a játra. Ledviny jsou výrazně zvětšeny a jsou prostoupeny malými cystami napříč dření i kortexem, mikroskopicky jsou charakterizovány dilatovanými sběrnými kanálky. Jaterní postižení vede k progresivní jaterní fibróze.
Patogeneze: v genu PKHD1 rozlišujeme až 300 mutací, ale i stejná mutace může mít rozličný průběh v konkrétní rodině. Nicméně existuje silná genotypově-fenotypová korelace. Mutace, která modifikuje fibrocystin a způsobuje lehčí postižení než mutace, které vedou ke zkrácení samotného proteinu.
Klinika: prenatálně jsou známky bilaterálního zvětšení ledvin s přítomností oligohydramnionu a přítomnost sekvence Potterové (facies Potteri): nízce nasedající uši, mikrognatie, oploštělý nos, abnormální ušní boltce, dále je přítomen IUGR a hypoplasie plic. Přítomnost oligurie a AKI jsou vzácné, iniciálně normální renální funkce má 20-30% pacientů. Formy, které se prezentují peri-neonatálně se vyvíjí v end stage renal disease (ESRD) zhruba do 10 let věku pacienta. Pokud se klinika objevuje v adolescenci, potom nejčastěji pod obrazem postižení ledvin a jater současně. Hepatální fibróza se manifestuje portální hypertenzí s jícnovými varixy, hepatosplenomegalií s hypersplenismem, caput medusae, ascendentní cholangoitida. Kongenitální jaterní fibrosa se může manifestovat dilatovanými intrahepatálními žlučovými cestami jako Caroli syndrome s rizikem bakteriálních cholangoitid.
Diagnóza: je založena na oboustranném zvětšení ledvin u dítěte s oligohydramniem a plicní hypoplasií, na ultrazvuku nacházíme hyperechogenní zvětšené ledviny se špatnou diferenciací mezi kůrou a dření ledvin společně s nálezem mikrocyst velikosti zhruba 1 mm. Diagnosu podporuje nález jaterní fibrosy, charakteristické jsou změny v jaterní biopsii. U rodičů nenacházíme známky onemocnění, u sourozenců je to možné. Potvrzení diagnózy je molekulárně-genetické.
Terapie: je pouze podpůrná, pokud není indikace k Tx játra - ledvina. Iniciálně agresivní ventilační podpora, ACEi a/nebo ARBs při hypertenzi a současné proteinurii. Pokud jsou ledviny zvětšeny do té míry, že způsobují respirační selhání a nebo intoleranci stravy pro vysoký stav bránice - indikace k nefrektomii (častěji oboustranné).
MUDr. Jiří Havránek, 8/2024
Galerie[upravit | editovat zdroj]
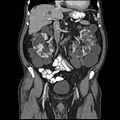
CT polycystické choroby ledvin

MRI obraz polycystické choroby ledvin
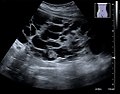
USG zobrazení polycystické ledviny



Souhrnné video[upravit | editovat zdroj]
Odkazy[upravit | editovat zdroj]
Použitá literatura[upravit | editovat zdroj]
- DÍTĚ, P., et al. Vnitřní lékařství. 2. vydání. Praha : Galén, 2007. ISBN 978-80-7262-496-6.