Porfyrie kožní


Porfyria cutanea tarda (PCT)[upravit | editovat zdroj]
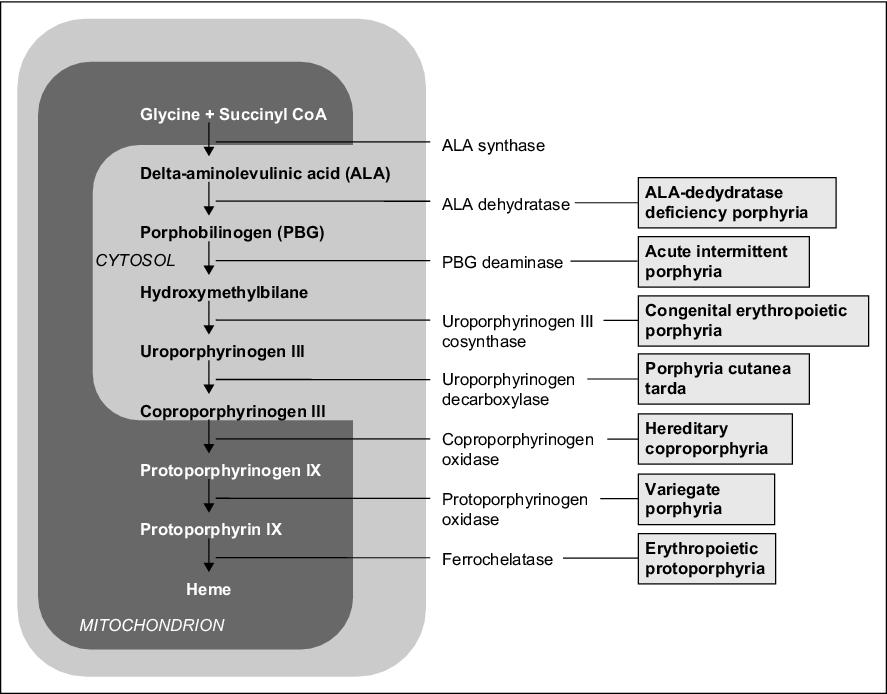
Jedná se o AD dědičný defekt uroporfyrinogendekarboxylasy, vyskytující se v poměru 1:25 000 (nejčastější forma), především u mužů středního věku. Porfyriny jsou v nadbytku vytvářeny v játrech, hromadí se zde, přenáší se krevním oběhem až do kůže, kde způsobují fotosenzitivitu, což je typický symptom. Po vystavení kůže slunečnímu záření se objevují tekutinou naplněné rozsáhlé puchýře, které se hojí velice pomalu za vzniku jizev a milií (tečkovitá bělavá ložiska). Kůže je hyperpigmentovaná, později atrofická, snadno zranitelná. Objevuje se hypertrichóza na spáncích a kolem očí. Klinická manifestace je dávána do souvislosti s poškozením jater způsobeným alkoholem, polyhalogenovanými uhlovodíky (hexachlorbenzen, dioxin), léčbou estrogeny, hepatomy, hemochromatózou nebo hepatitidou. Neléčená může vést ke karcinomu jater. Existuje i nedědičná forma (sporadická, tzv. PCT 1. typu). V moči nacházíme uroporfyrin, vysokou hladinu železa, v 50 % případů vysoké hladiny jaterních enzymů.
Léčba: opakované venepunkce (300−500 ml ve 2−4 týdenním intervalech) zbavující tělo nadbytečných porfyrinů a železa + podávání antimalarika chlorochininu (125-250 mg denně), které způsobuje pomalé vyplavování porfyrinů, dále pak ochrana před slunečním zářením (oděv, speciální krémy) a jaterní dieta.
Vrozená (kongenitální) erytropoetická porfyrie (CEP, Güntherova choroba)[upravit | editovat zdroj]
Jedná se o AR dědičný defekt uroporfyrinogen-III-synthasy (UROS) vedoucí k zvýšené tvorbě porfyrinů v kostní dřeni, které se hromadí v organismu, hlavně v erytrocytech. Výskyt je 1:2−3 mil. Tato choroba se zpravidla projeví již v dětství. Projevy onemocnění se různí – patří mezi ně např. tmavě červená moč (dáno přítomností uroporfyrinu a koproporfyrinu), citlivost kůže (tvorba puchýřů, jizvení) a její tmavnutí, citlivost očí, ztráta řas, anémie, splenomegalie, červené zbarvení zubů, nadměrné ochlupení (zvláště na rukou a v obličeji).
Léčba: transplantace kostní dřeně, ochrana před slunečním zářením, krevní transfuze, splenektomie.
.jpg)
Protoporfyrie (EPP)[upravit | editovat zdroj]
Jedná se o AD dědičný defekt ferrochelatasy, jehož následkem dochází k hromadění protoporfyrinu v játrech, kostní dřeni a kůži. Nejčastějšími příznaky jsou zarudnutí, svědění a otok kůže i po krátkodobém (několik minut) vystavení kůže slunečnímu záření. Příznaky vymizí po hodinách až dnech, při opakovaném vystavení dochází k jizvení kůže a dalším variabilním kožním projevům. Choroba se většinou projeví již v dětství. V několika málo procentech případů dochází k jaternímu poškození.
Léčba: zmírnění projevů pomocí beta-karotenu, antihistaminik, melanotanu, fototerapie; prevencí je používání ochranného oblečení a speciálních krémů. Na rozdíl od akutních jaterních porfyrií EPP nezhoršují žádné léky.
{kind=link}