Porfyrie
(přesměrováno z Porfyriny)



Porfyrie jsou skupina metabolických onemocnění s poruchou syntézy hemu, vyznačují se akumulací nebo zvýšenou exkrecí porfyrinů. Porfyrie můžeme dělit na erytropoetické, hepatální a erytrohepatální. Nejčastější ze skupiny těchto onemocnění je porphyria cutanea tarda.[1]

Repetitorium biochemie – porfyriny
Porfyriny jsou cyklické sloučeniny tvořené čtyřmi pyrrolovými kruhy spojenými methinovými (=CH-) a u porfyrinogenů methylenovými můstky (-CH2-). Následně tvoří komplexy s ionty kovů (např. Fe v hemoglobinu, myoglobinu, cytochromech, katalase; Co ve vitaminu B12; Mg v chlorofylu).
Jsou barevné v různých odstínech červeně (na rozdíl od bezbarvých porfyrinogenů). Zbarvení je dáno systémem konjugovaných dvojných vazeb v jejich molekule, které absorbují viditelné světlo. V UV světle červeně fluoreskují. Z důvodu nepostradatelnosti pro život se musí v těle de novo syntetizovat (přijímané v potravě se rozkládají a slouží jako zdroj živin).
Charakteristika porfyrií
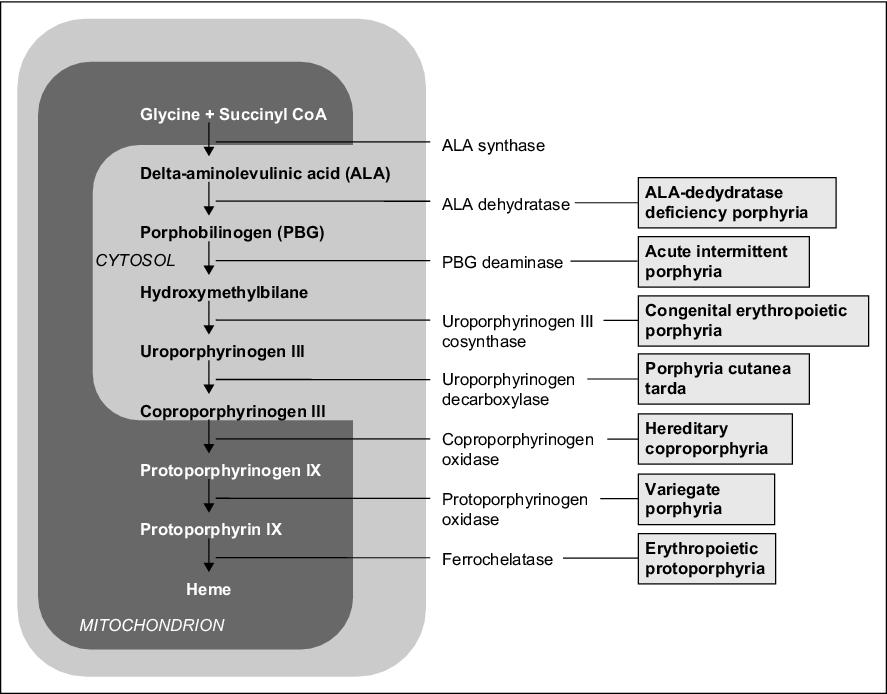
Mutacemi genů, které řídí syntézu enzymů působících při syntéze hemu, dochází k nedostatku produktu metabolismu za blokem (hemu) nebo nahromadění metabolitu před ním.
Při defektu enzymu v časné fázi biosyntézy (před tvorbou porfyrinogenů) se v tělesných tekutinách hromadí výchozí látky ALA – kyselina δ-aminolevulová (kyselina 5-aminolevulová) a PBG – porfobilinogen, které mají toxický účinek (interferují se synaptickými funkcemi) na PNS a CNS, čímž vznikají typické příznaky – neuropatie (svalová slabost), abdominální bolest, zvýšená aktivita sympatiku a neuropsychické problémy (neklid, hysterie, psychotické stavy).
Defekty ve vyšších stupních syntézy vedou k hromadění porfyrinogenů, jejichž oxidační produkty (odpovídající porfyriny) způsobují fotosenzitivitu (hyperreakci na viditelné světlo v oblasti 400 nm). Při vystavení porfyrinů světlu o této vlnové délce dochází k jejich excitaci, reakci s molekulárním kyslíkem za tvorby kyslíkových radikálů, které poškozují buněčné organely včetně lysosomů → dochází k uvolnění lysosomálních enzymů, které poškozují světlu vystavenou kůži (erytém, puchýře, jizvení). Mezi příznaky onemocnění patří také červenání až hnědnutí zubů, moči a stolice, případně ústup dásní.
Jednotlivé způsoby eliminace mají význam v diagnostice porfýrií. ALA, PBG, uroporfyrin jsou ve vodě rozpustné → vylučují se močí. Protoporfyrin není rozpustný ve vodě → vylučuje se žlučí. Koproporfyrin se nachází ve žluči i moči, přičemž jeho vylučování močí stoupá při poškození jater.
Manifestace bývá často až v dospělosti po určitém vyvolávajícím momentu (léky, oslunění, stres, hormonální vlivy). Podáním některých léků (barbituráty, anestetika) a také alkoholu se zvyšuje aktivita ALA-synthasy, což vede ke zvýšené tvorbě porfyrinů, a tím zhoršení příznaků u pacientů s porfyriemi (eventuálně akutní atace). Akutními porfyriemi jsou častěji postiženy ženy, chronickými muži.
V akutní terapii glukóza indukci ALA-syntasy potlačuje, účinek se potencuje aplikací derivátů hemu jako hem-albumin nebo hem-arginát nebo hematin. Hem inhibuje negativní zpětnou vaznou ALA-syntasu.[2]

Dělí se podle:
- nejvíce postižených orgánů produkujících porfyriny:
- hepatální – AIP, PCT, HCP, VP, ADP;
- erytropoetické – CEP;
- erytrohepatální – EPP;
- projevů:
- kožní – PCT, EPP, CEP; VP a HCP také způsobují kožní projevy;
- jaterní – AIP, ADP, VP, HCP;
- průběhu:
- akutní – AIP, VP, HCP, ADP;
- chronické – PCT, EPP, CEP;
| Typ porfyrie | Postižený enzym | Hlavní příznaky | Laboratorní nález |
| Akutní intermitentní (hepatální) | uroporfyrinogen I kosynthetasa | abdominální bolest, neuropsychiatr. příznaky, není fotosenzitivita | U-PBG ↑,
U-uroporfyrin ↑ |
| Kongenitální erytropoetická | uroporfyrinogen III kosynthetasa | fotosenzitivita | U-uroporfyrin ↑,
U-PBG ↓ |
| Porphyria cutanea tarda (kožní) | uroporfyrinogen dekarboxylasa | fotosenzitivita | U-uroporfyrin ↑ |
| Porphyria variegata (hepatální) | protoporfyrinogenoxidasa | fotosenzitivita, abdominální bolest, neuropsychiatrické příznaky | U-PBG ↑,
F-protoporfyrin ↑ |
| Protoporhyria (erytrohepatální) | ferrochelatasa | fotosenzitivita | F-protoporfyrin ↑,
Ery-protoporfyrin ↑ |
| Hereditární koproporfyrie (hepatální) | koproporfyrinogenoxidasa | fotosenzitivita, abdominální bolest, neuropsychiatrické příznaky | U-PBG ↑,
U-uroporfyrin ↑ |
Porfyrie kožní
Porfyria cutanea tarda (PCT)
Jedná se o AD dědičný defekt uroporfyrinogendekarboxylasy, vyskytující se v poměru 1:25 000 (nejčastější forma), především u mužů středního věku. Porfyriny jsou v nadbytku vytvářeny v játrech, hromadí se zde, přenáší se krevním oběhem až do kůže, kde způsobují fotosenzitivitu, což je typický symptom. Po vystavení kůže slunečnímu záření se objevují tekutinou naplněné rozsáhlé puchýře, které se hojí velice pomalu za vzniku jizev a milií (tečkovitá bělavá ložiska). Kůže je hyperpigmentovaná, později atrofická, snadno zranitelná. Objevuje se hypertrichóza na spáncích a kolem očí. Klinická manifestace je dávána do souvislosti s poškozením jater způsobeným alkoholem, polyhalogenovanými uhlovodíky (hexachlorbenzen, dioxin), léčbou estrogeny, hepatomy, hemochromatózou nebo hepatitidou. Neléčená může vést ke karcinomu jater. Existuje i nedědičná forma (sporadická, tzv. PCT 1. typu). V moči nacházíme uroporfyrin, vysokou hladinu železa, v 50 % případů vysoké hladiny jaterních enzymů.
Léčba: opakované venepunkce (300−500 ml ve 2−4 týdenním intervalech) zbavující tělo nadbytečných porfyrinů a železa + podávání antimalarika chlorochininu (125-250 mg denně), které způsobuje pomalé vyplavování porfyrinů, dále pak ochrana před slunečním zářením (oděv, speciální krémy) a jaterní dieta.
Vrozená (kongenitální) erytropoetická porfyrie (CEP, Güntherova choroba)
Jedná se o AR dědičný defekt uroporfyrinogen-III-synthasy (UROS) vedoucí k zvýšené tvorbě porfyrinů v kostní dřeni, které se hromadí v organismu, hlavně v erytrocytech. Výskyt je 1:2−3 mil. Tato choroba se zpravidla projeví již v dětství. Projevy onemocnění se různí – patří mezi ně např. tmavě červená moč (dáno přítomností uroporfyrinu a koproporfyrinu), citlivost kůže (tvorba puchýřů, jizvení) a její tmavnutí, citlivost očí, ztráta řas, anémie, splenomegalie, červené zbarvení zubů, nadměrné ochlupení (zvláště na rukou a v obličeji).
Léčba: transplantace kostní dřeně, ochrana před slunečním zářením, krevní transfuze, splenektomie.
.jpg)
Protoporfyrie (EPP)
Jedná se o AD dědičný defekt ferrochelatasy, jehož následkem dochází k hromadění protoporfyrinu v játrech, kostní dřeni a kůži. Nejčastějšími příznaky jsou zarudnutí, svědění a otok kůže i po krátkodobém (několik minut) vystavení kůže slunečnímu záření. Příznaky vymizí po hodinách až dnech, při opakovaném vystavení dochází k jizvení kůže a dalším variabilním kožním projevům. Choroba se většinou projeví již v dětství. V několika málo procentech případů dochází k jaternímu poškození.
Léčba: zmírnění projevů pomocí beta-karotenu, antihistaminik, melanotanu, fototerapie; prevencí je používání ochranného oblečení a speciálních krémů. Na rozdíl od akutních jaterních porfyrií EPP nezhoršují žádné léky.
Porfyrie jaterní
Akutní intermitentní porfyrie (AIP)
Podkladem pro vznik je AD dědičný defekt hydroxymetylbilan syntázy (jinými názvy porfobilinogen deamináza, PBGD nebo uroporfyrinogen-I-syntáza) vedoucí k hromadění prekurzorů hemu v játrech. Manifestuje se akutní atakou po zátěži některými chemickými látkami (steroidy, léky, alkohol), hladověním, infekcí či stresem; většinou v období po pubertě. Hlavní příznaky jsou abdominální bolest (imitující NPB), zácpa, zvracení, hypertenze a psychické problémy (hysterie), bolesti hlavy, parézy až plegie. V moči se nachází zvýšená hladina ALA a PBG. V krvi dominuje hyponatrémie, hypokalémie s abnormalitami metabolismu cukrů a tuků. Diagnózu potvrdí snížená aktivita PBGD v erytrocytech.
Léčba: v akutní fázi infuze s glukózou (inhibuje ALA-syntázu) a hematinem; prevencí další ataky je vyvarování se vyvolávající látky (zákaz užívání některých léčiv nebo alkoholu).
Porfyrie z deficitu 5-aminolevulátdehydratázy (ADP, Dossova porfyrie)
Způsobuje ji AR dědičný deficit 5-aminolevulátdehydratázy.
Symptomy jsou abdominální bolesti a neuropsychické potíže. V moči je přítomná ALA a koproporfyrin.
Hereditární koproporfyrie (HCP)
Vzniká AD dědičným defektem koproporfyrinogenoxidázy.
Symptomy zahrnují neuropsychické potíže, fotosenzitivitu, vzácně abdominální bolest, avšak časté jsou i naprosto asymptomatické formy. V akutním stádiu jsou v moči zvýšené hladiny ALA, PBG, koproporfyrinu (ten je také prokazatelný ve stolici).
Porfyria variegata (VP)
Etiologie je AD dědičný defekt protoporfyrinogenoxidázy.
Symptomy jsou abdominální bolest, neuropsychické potíže a u některých postižených také kožní příznaky (fotosenzitivita). V moči prokazatelné vysoké hodnoty ALA, PBG, koproporfyrinu; ve stolici zvýšená exkrece protoporfyrinu a koproporfyrinu.
Diagnóza
Stavenovením porfyrinů v tělesných tekutinách, v moči a ve stolici (specifická fluorescence).
Pozn.: porfyrinurie a vylučování ALA mohou být ale také příznakem při otravě olovem (inhibuje ALA-dehydratasu a ferrochelatasu), jehož výsledkem je anémie a nedostatek ATP.
Léčba porfyrií
Nemocní by měli dbát na správnou životosprávu – dostatek vitaminů; vyvarovat se alkoholu, česneku (obsahuje enzymy zhoršující projevy porfyrie), slunečního světla a UV záření. K symptomatologické léčbě jsou využívány krevní transfuze a vstřikování hemu.
Odkazy
Související články
Externí odkazy
{kind=link}
Zdroj
- MASOPUST, Jaroslav a Richard PRŮŠA. Patobiochemie metabolických drah. 2. vydání. Univerzita Karlova, 2004. 208 s. s. 118–119.
Reference
- ↑ ČEŠKA, Richard, et al. Interna. 1. vydání. Praha : Triton, 2010. 855 s. s. 315-317. ISBN 978-80-7387-423-0.
- ↑ KÖNIGSHOFF, Melanie, et al. Kurzlehrbuch Biochemie. 3. vydání. Thieme, 2012. 406 s. s. 276. ISBN 9783131364135.
Použitá literatura
- MURRAY, Robert, Daryl GRANNER a Peter MAYES, et al. Harperova biochemie. 4. české vydání. Jinočany : Nakladatelství H+H, 2002. 872 s. s. 354-360. ISBN 80-7319-013-3.
- LEDVINA, Miroslav, Alena STOKLASOVÁ a Jaroslav CERMAN. Biochemie pro studující medicíny : II.díl. 2. vydání. Praha : Nakladatelství Karolinum, 2009. s. 336-341. ISBN 978-80-246-1415-1.
- MASOPUST, Jaroslav a Richard PRŮŠA. Patobiochemie metabolických drah. 1. vydání. Praha : Univerzita Karlova, 2. lékařská fakulta, 1999. 182 s. s. 104-110. ISBN 80-238-4589-6.
- KALOUSOVÁ, Marta, et al. Patobiochemie ve schématech. 1. vydání. Praha : Grada Publishing a.s, 2006. 264 s. s. 51-57. ISBN 80-247-1522-8.